From Bisulfite Chemistry to Single Cell Methylation Maps
Single-cell bisulfite sequencing is a laboratory method that lets scientists read DNA methylation, a chemical mark that can turn genes on or off, one cell at a time. Bisulfite treatment converts unmethylated cytosines into another base while leaving methylated cytosines unchanged. Sequencing then reveals which sites were methylated across the genome. At single-cell resolution, this generates extremely high-dimensional data: millions of possible methylation sites, but only a tiny fraction observed in each cell due to limited DNA and sequencing depth. The result is a matrix that is mostly missing values, riddled with technical noise and biases introduced by library preparation and alignment tools. Yet buried inside are subtle epigenomic patterns that distinguish cell subtypes, developmental states and disease processes, from cancer to neurodegeneration. Making sense of this sparse, noisy landscape requires more than traditional statistics; it calls for AI in life sciences that can model uncertainty and structure directly.
How the MethylVI Deep Generative Model Cleans Up the Noise
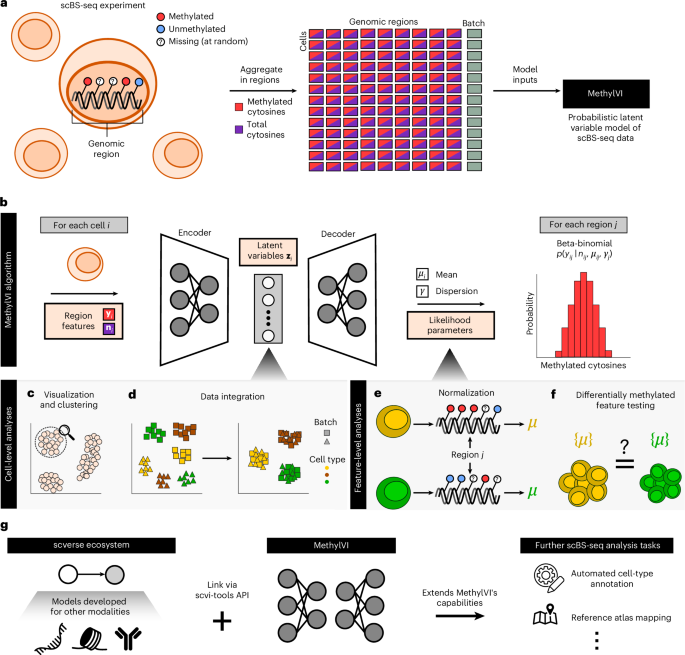
MethylVI is a probabilistic deep generative model designed specifically for single cell methylation data. Instead of treating every missing or noisy measurement as a simple zero, it learns a low-dimensional representation—the latent space—that captures key biological factors such as cell type and state, while explicitly modelling technical variability. Conceptually, the model assumes that each cell’s true methylation profile is generated from a small set of hidden variables, then distorted by experimental noise from bisulfite conversion, sequencing and alignment. By training on large datasets, MethylVI learns this generative process and can infer denoised methylation patterns, impute unobserved sites and correct batch effects. This improves downstream bioinformatics data analysis tasks, such as clustering cells, identifying differentially methylated regions and integrating methylation with other single-cell omics. In practice, MethylVI acts as an AI-powered filter: it preserves meaningful epigenetic signals while down-weighting artefacts that would mislead simpler tools.
AI in Life Sciences: From Models Like MethylVI to Market Momentum
MethylVI sits within a fast-expanding ecosystem of epigenomics AI tools and life sciences analytics platforms. Market analyses of AI in life sciences highlight drug discovery, diagnostics and personalised medicine as primary growth engines, with major players including technology vendors, medical imaging specialists and bioinformatics-focused companies. AI-driven platforms are already shortening drug discovery timelines by analysing vast, heterogeneous datasets and prioritising promising targets more quickly. In hospitals, predictive analytics tools are helping clinicians anticipate complications and reduce readmission rates, illustrating how similar techniques could be applied to methylation-informed risk scores. As precision medicine gains ground, models that can interpret single cell methylation data will be crucial for tailoring treatments based on a patient’s molecular profile. Probabilistic deep generative models, GPU-accelerated pipelines and cloud-hosted software stacks are turning once esoteric epigenomic datasets into routine analytes that fit directly into commercial and clinical decision workflows.
Practical Payoffs: Finding Cell Subtypes, Signatures and Targets Faster
In concrete terms, AI-powered analysis of single cell methylation can accelerate discoveries that matter at the bedside. Deep generative models enable more accurate clustering of cells, revealing previously hidden subtypes in tissues such as brain or tumour microenvironments. These subtypes may carry distinct epigenetic signatures—patterns of methylation at specific genomic regions—linked to disease progression, treatment resistance or adverse outcomes. Instead of manually scanning thousands of loci, researchers can use AI to prioritise regions whose methylation shifts consistently across patients, nominating them as potential biomarkers or therapeutic targets. Integrating methylation with gene expression and 3D genome architecture further refines this view, helping to map regulatory elements that could be modulated by drugs. Compared with traditional pipeline-style analysis, AI tools can explore larger search spaces, quantify uncertainty and generate ranked hypotheses, allowing labs, hospitals and biotech startups to move from raw reads to actionable insights more rapidly.
Infrastructure, Regional Opportunity and Ethics in the Genomic AI Era
Unlocking the value of models like MethylVI requires robust data infrastructure: scalable storage for large sequencing files, GPU compute for training and running deep networks, and specialised bioinformatics talent to design and validate pipelines. Cloud-based platforms are lowering these barriers, making advanced epigenomics AI tools accessible beyond top Western institutes and opening doors for universities, hospitals and biotech startups in Malaysia and the wider region. Local teams can upload raw single-cell bisulfite data, run state-of-the-art models and focus on interpreting results for regional disease burdens, such as cancer and metabolic disorders. However, applying AI in life sciences to genomic and patient-linked methylation data raises serious ethical and regulatory issues. Data governance must address consent, anonymisation, cross-border transfers and algorithmic bias. Even at the analysis stage, clear oversight, transparent methods and alignment with emerging health data regulations are essential to ensure that powerful new models serve patients fairly and securely.